Marfan syndrome is inherited through an autosomal dominant mutation of the gene encoding glycoprotein fibrillin-1 (FBN1), which plays a role in the anchoring of cells to the extra-cellular matrix and is the main component of microfibrils . Microfibrils affect the strength and elasticity of connective tissue, and also control the release of growth factors that cause growth and repair of tissues and organs throughout the body. A defect in this gene means that the quality and/or quantity of fibrillin-1 in the body is decreased, reducing the amount available to form microfibrils, implicating changes in the body’s connective tissues and growth of tissues. Marfan’s most widely known physical characteristics include a tall, thin build with long fingers, arms, and legs.
Characteristics/Clinical Presentation
Musculoskeletal System
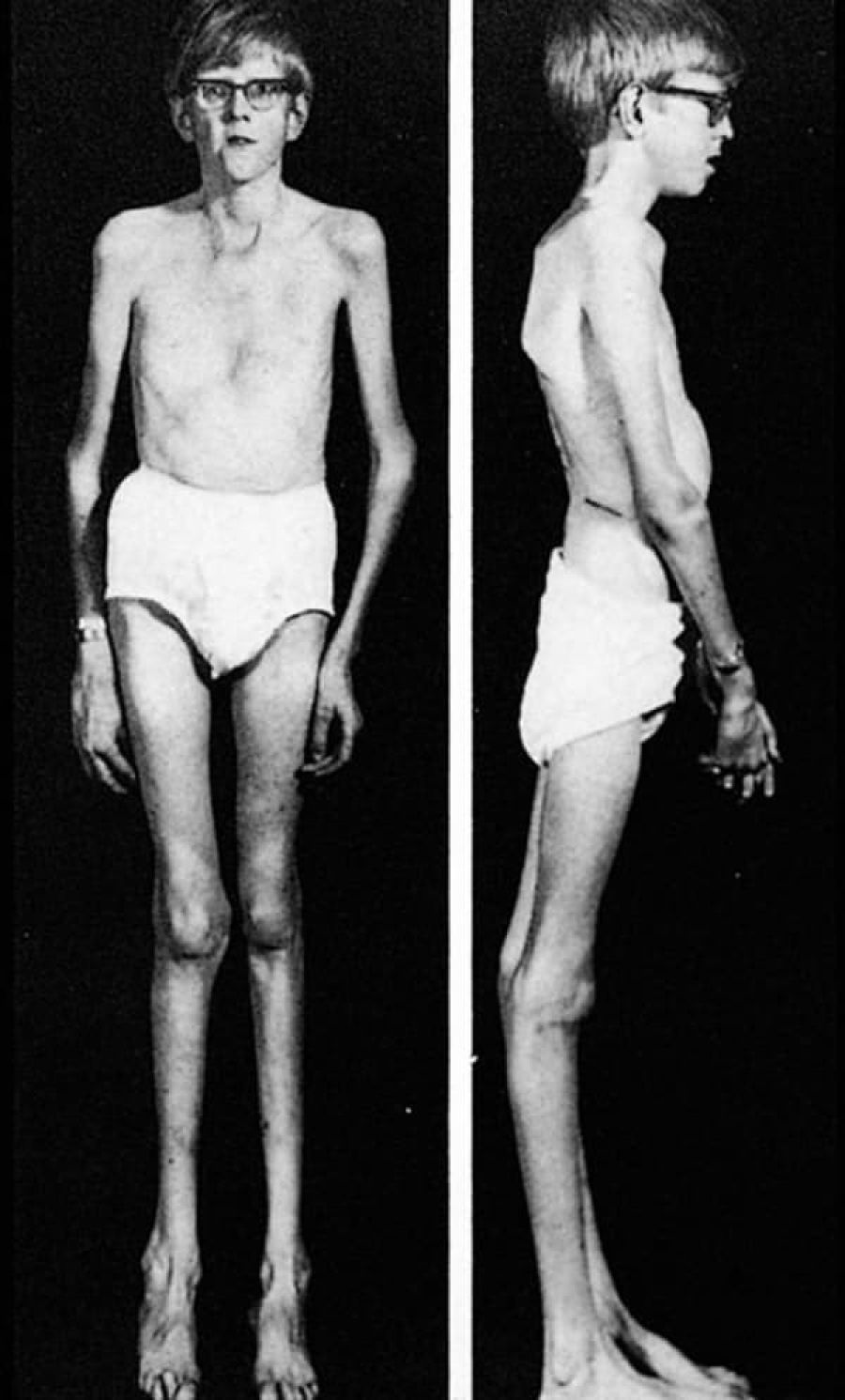
People with Marfan syndrome can display any or all of the following physical characteristics: tall, thin body build; long arms and legs (dolichostenomelia); elongated fingers and toes (arachnodactyly); unusually flexible joints; long narrow face; highly arched roof of the mouth; crowded teeth; small lower jaw; scoliosis; pes planus; pectus excavatum; pectus carinatum; and arm span that exceeds body height.
Ocular System
It is very common for people with Marfan syndrome to be myopic. Other ocular symptoms include early glaucoma and cataracts. More than half of those with Marfan syndrome will also experience dislocation of one or both lenses of the eye and possibly retinal detachment.
Cardiovascular System
Patients with Marfan syndrome are at risk for a variety of cardiac problems, including: aortic root dilation; aortic valve regurgitation; aortic dissection, aneurysm, or rupture; mitral valve prolapse; bacterial endocarditis; cardiomyopathy; heart murmur; intracranial bleeding/Berry aneurysm; and heart failure. A patient with these complications may complain of shortness of air, heart palpitations, and fatigue.
Nervous System
Patients with Marfan syndrome are at risk for dural ectasia later in life, which can be a source of discomfort or pain including low back pain, radiating pain/numbness to the abdomen or legs, bowel and/or bladder changes, and headache.
Pulmonary System
Patients with Marfan syndrome are at risk for recurrent spontaneous pneumothorax which can cause sudden pain (especially on one side of the chest) and/or shortness of air. These patients may also snore or experience sleep apnea. They are also at an increased risk for asthma, pneumonia, bronchitis, emphysema, and cystic lung disease due to the changes in the lung tissue that occur with Marfan syndrome.
Integumentary System
Stretch marks that are unexplained by weight gain/loss or pregnancy are common in patients with Marfan syndrome. They are also at an increased risk of developing hernias, especially abdominal or inguinal.
Associated Co-morbidities
The section above regarding Characteristics/Clinical Presentation lists many of the co-morbidities associated with Marfan syndrome.
Medications
There is no cure for Marfan syndrome, and as such medications are used on a case-by-case basis based on the patient’s individual needs:
β-blockers, ACE-inhibitors, calcium channel blockers, or angiotensin II receptor blockers: used to reduce cardiovascular complications such as dissecting aorta by reducing myocardial contractility and pulse pressure
Estrogen and progesterone: sometimes used to induce precocious puberty in very tall girls before age 10 to reduce potential adult height.
Prophylaxis for bacterial endocarditis: antibiotics given before invasive dental or GI procedures to reduce the risk of introduction of bacteria to the blood stream
Pain medications: used to relieve pain associated with various musculoskeletal “aches and pains” as well as from dural ectasia.
Diagnostic Tests/Lab Tests/Lab Values
Diagnosis of Marfan syndrome is based on the clinical criteria (see below), and may be assisted by echocardiogram/electrocardiogram/MRI for measurement of the aortic root and detection of valve prolapse, slit-lamp examination for the assessment of lens abnormalities, x-ray studies of the skeletal system for assessment of characteristic abnormalities, and MRI to assess for dural ectasia. Genetic testing is also available for assistance in the diagnosis of Marfan syndrome, although clinical findings and genetic/family history are currently the most effective methods of diagnosis.
Etiology/Causes
Marfan syndrome is caused by a genetic mutation of the fibrillin-1(FBN1) gene. This defect can be inherited in an autosomal dominant manner from a parent, or can happen spontaneously at conception. In spontaneous cases, the cause of the genetic mutation is unknown.
Systemic Involvement
As a syndrome, Marfan’s has many implications on the various body systems. Systemic Involvement associated with Marfan syndrome has been laid out in the Characteristics/Clinical Presentation section above. However, almost every patient with Marfan syndrome will experience musculoskeletal symptoms and nearly 80% experience cardiovascular symptoms, so it would be prudent to expect these body systems to be implicated in a patient with Marfan syndrome.
Medical Management
Medical management for Marfan syndrome is individualized for each patient based on their specific clinical manifestations. This often includes preventative measures such as medications or regular diagnostic procedures, as well as surgery.
Regular checkups with a cardiologist including routine echocardiograms are recommended for patients with Marfan syndrome to evaluate the size of the aorta and general heart function. Aortic valve repair or replacement surgeries are often performed once the aorta reaches a size that shows a high risk for tear or rupture—generally 4.7-5cm, depending on the height of the individual. Surgery is performed preventatively because the risk of death once the aorta dissects is approximately 40%, and even after emergency surgery the mortality rate is 10-20%.
Patients with Marfan syndrome may need the attention of an orthopedic physician to address their musculoskeletal symptoms, including scoliosis, pectus excavatum, and pectus carinatum. These deformities can sometimes lead to infringement of chest expansion causing breathing difficulties, as well as other problems like pain and cosmetic concerns. Some of these conditions may require bracing or surgery.
Management by an optometrist or ophthalmologist and regular eye exams are recommended for the patient with Marfan syndrome due to the ocular issues that can occur with this disease. Some problems, like myopia or detached lenses, can be solved with only eyeglasses, contact lenses, or intraocular lens implants if desired. Other issues like glaucoma, cataracts, and detached retina may be managed with eye drops, oral medications, or surgery.
Women with Marfan syndrome who wish to become pregnant should be under the medical management of an obstetrician due to the increased strain and even higher risk to the cardiovascular system during pregnancy and birth. Genetic counselling may also be helpful to the mother regarding the likelihood of passing her or her partner’s genetic disorder to their child.
Patients with Marfan syndrome may benefit from the medical management of other professionals such as: dentist/orthodontist for dental/cosmetic issues; podiatrist for problems associated with pes planus; neurologist for problems associated with dural ectasia; and pulmonologist for recurrent spontaneous pneumothorax, sleep apnea, and other lung problems.
Physical Therapy Management
Intervention
There are no specific protocols used by physical therapists in the management of Marfan syndrome. Because patients with Marfan syndrome have different clinical manifestations, physical therapy may be helpful to the individuals who experience problems from the musculoskeletal issues that are often associated with Marfan syndrome. This includes but is not limited to pes planus, scoliosis, and pectus excavatum and pectus carinatum and the physical issues they may cause. Stretching, strengthening, therapeutic exercises, modalities for pain, fabricating or recommending orthotics, as well as patient education are all valuable tools the physical therapist can use in the management of the patient with Marfan syndrome. Physical therapists can also play a role in advising the patient with Marfan syndrome on the risks and benefits of exercise and participation in sports. Specifically, the patient should be educated on the risk to the cardiovascular system with strenuous and contact sports and should be advised on the National Marfan Foundation’s Physical Activity Guidelines, prepared by the NMF Professional Advisory Board.